Scaling Screening Across 36 Billion Compounds On-Prem 🌐🚀
10 August 2023
After Recursion acquired Cyclica in May 2023, the company set a bold target: to screen the single largest virtual compound library to date. I was on the team that turned that goal into reality.

We leveraged BioHive-1’s 63‑DGX H100 to benchmark GPU configurations and optimize our MatchMaker inference, ultimately enabling us to rapidly predict drug–target interactions across Enamine’s REAL Space library at an unprecedented scale.
At the heart of this effort is MatchMaker: a customized neural network toolkit trained on 4 million drug-target interaction pairs, meticulously sourced from comprehensive bioactivity repositories. These DTIs are mapped onto protein 3D binding pockets and further enriched with structural data, extracted from experimental sources or predicted via AlphaFold, stored in our internal PSSB database. To me, MatchMaker represents a powerful tool: coupling an extended DTI dataset with structural biology to bridge computational prediction and real-world binding relevance.
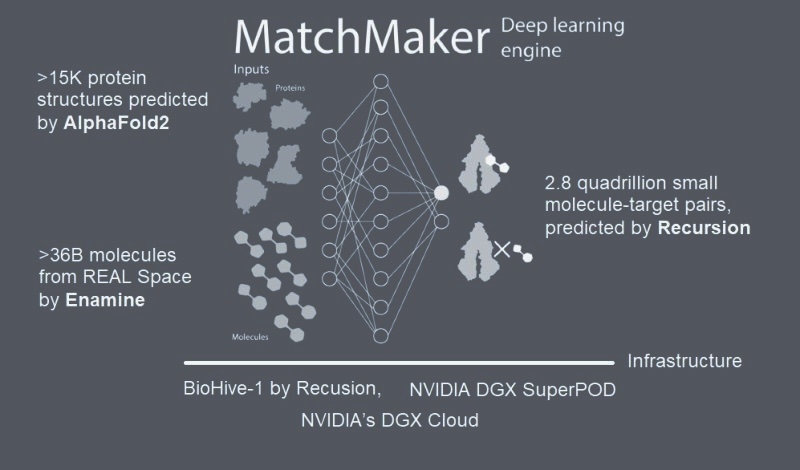
A Screen at Unmatched Scale
With this system in place, we screened 36 billion molecules from Enamine Ltd. against 15,000 human proteins. This covers more than 80,000 potential binding pockets! In total, this project aimed to screen over 2.8 quadrillion protein–ligand pairs. This is a milestone in drug discovery not just for its size, but for what it enables:
- Enhanced Targeting: We can now prioritize ligands for nearly any human protein, setting up higher-resolution follow-ups using docking or simulations.
- Improved Selectivity Forecasting: The ability to predict off-target interactions allows for early elimination of problematic compounds, hence helping reduce late-stage failures.
- Smarter Hit Triage: With massive hit lists for every target, teams can filter using ADMET or other heuristics to select only the most viable candidates.
- Faster FBDD: Predicted binders can be fragmented for use in Fragment-Based Drug Discovery pipelines, reducing synthetic overhead.
This level of scale and structural insight radically simplifies and accelerates many steps that traditionally required slow, expensive trial-and-error.
Why this matters?
- Speed at scale. Our GPU-tuned MatchMaker runs orders of magnitude faster than docking tools, transforming how we prioritize compounds for experimental follow-up.
- Proteome‑wide reach. MatchMaker scans across the proteome, spotting opportunities that docking and chem‑centric screens might miss.
- From predictive to practical. Instead of sifting through billions of molecules, teams now get curated libraries that are not only promising computationally, but synthetically deliverable.
- We moved fast. Within 90 days of closing the Cyclica acquisition, and just 30 days after launching our NVIDIA collaboration, we generated this massive new data layer of predicted interactions.
My contribution
I was responsible for benchmarking and tuning MatchMaker inference workloads on Recursion’s BioHive. I tested mixed-precision configurations, optimized batch sizes, andmonitored memory usage to make sure MatchMaker could reliably screen the trillions of compounds we had in the Enamine database.
Working at the intersection of AI performance engineering and structure-based drug discovery showed me just how game-chaning the combination of compute and science can be. By bringing MatchMaker’s structural DTI insights to 36 billion Enamine molecules and running it reliably on Biohive-1, I helped power a key milestone in the Drug Discpvery pipeline at Recurison that closes the loop between virtual prediction and tangible, purchasable chemistry. This screen isn’t just about quantity: it’s about actionable, structure-aware prediction at a scale that changes what’s possible in early discovery. It’s a bold new chapter in discovery-driven biotech, and I’m proud to have been a part of it!