Membrane Curvature from MD Simulations: Considerations [Part I]
22 July 2021
Some points to keep in mind to calculate membrane curvature from MD simulations.
Molecular Dynamics(MD) simulations are a powerful tool to study biological systems at the molecular level of detail. Over the last decades, biomolecular systems investigated via MD simulations have gained an incredibly high complexity while extending to the microseconds and even milisecond time-scale. The remarkable flexibility to reproduce realistic biomolecular systems, together with the ability to perform analysis over long time scales while keeping track of physico-chemical interactions, makes of MD one of the most popular tools across different fields of science and engineering.

Lipid bilayers play a pivotal role in in biological systems. By providing specific biophysical properties, lipids confer structure and unique physical properties to the bilayer [1]. Such physical properties strongly relate to their biological function, and to make everything even more interesting, the composition of each cellular membrane has evolved to optimize the functions associated with it. This makes of MD simulations of lipid bilayers, in broad ranges of lipid composition and size, is one of the most common MD setups in biomolecular systems.
However, biological membranes are not purely a lipid barrier. They also contain up to 50 % protein by mass (up to 50 %!) [2] . Membrane proteins are critical components in the biology of the cell. Several studies support that interactions between proteins and their lipid environment may influence the stability and function. This makes of MD simulations of membrane-protein systems a very common approach.
MD simulations produce high volumes of data that also requires exhaustive analysis and statistics. Since our main goal is to develop a tool to calculate membrane curvature, we should look at the problem from different perspectives. In the first part of this series, I will describe some important points to consider when developing analysis tools to calculate membrane curvature from MD simulations.
Together with Numpy, the foundation of this cool MembraneCurvature tool is MDAnalysis.
In the following considerations I will use the names of key data structures and concepts from
MDAnalysis.
Considerations (Part I)
At the most fundamental escenario, we have two possible MD simulations setups:
1.1 Membrane only.
and
1.2 Membrane-protein.
Simultaneously, in membrane-protein systems, we can identify two types of setups:
1.2.1. Protein with positions restraints:
The protein is fixed: neither translates nor rotates.
1.2.2. Protein with no position restraints:
The protein diffuses in the lipid bilayer and is free to translate and rotates.
Figure 1 shows a diagram that summarizes the most common system setups in MD.
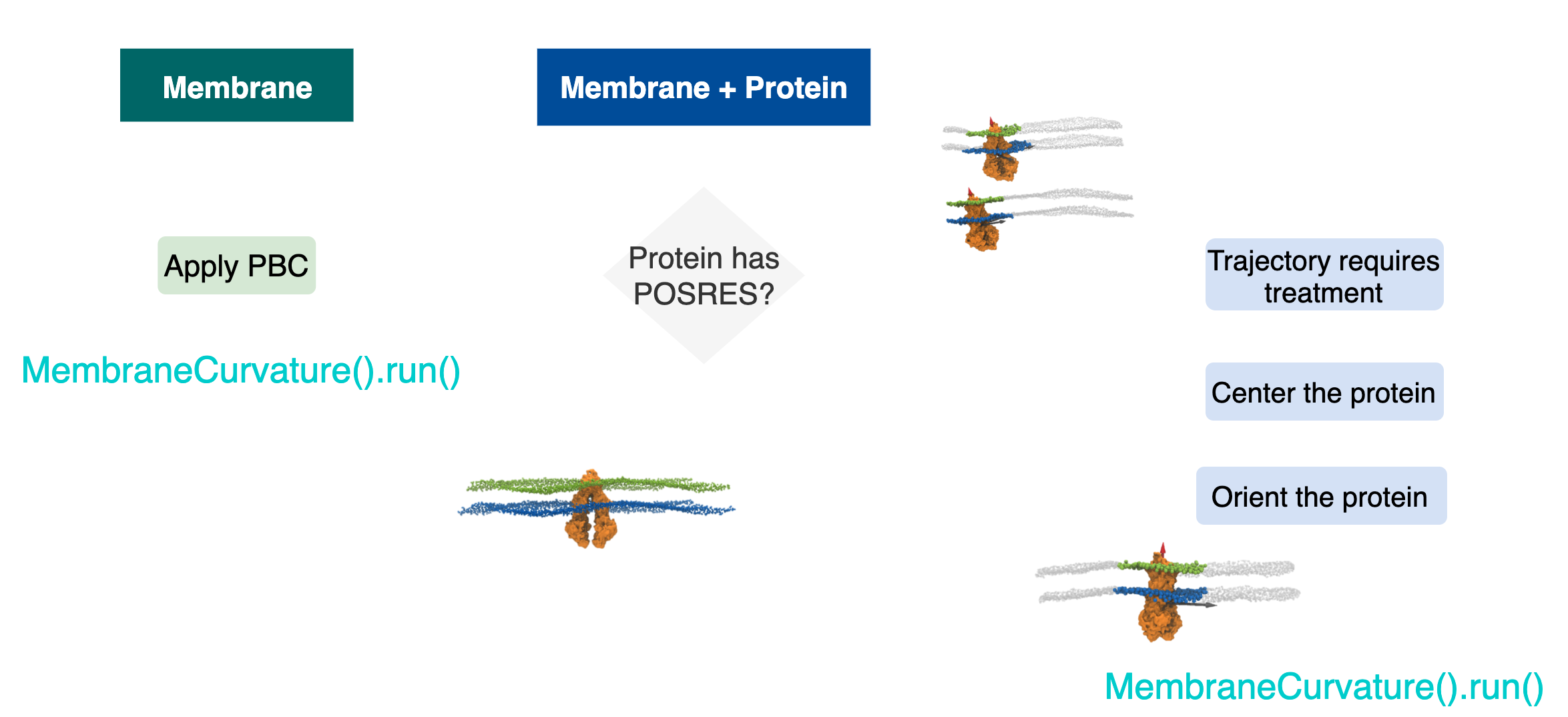
In one category, we can put together two types of setups: 1.1 Membrane only systems and 1.2.1 Membrane protein systems and Protein with positions restraints.
In both cases, we don’t need further trajectory processing. Instead, what we
need is to treat all the atoms that fall outside the boundaries of our
simulation box in the n_frames of the trajectory. We can manage these
jumpy atoms by using the MDAnalysis.transformations.wrap.
This is a schematic example of how wrapped coordinates look like:
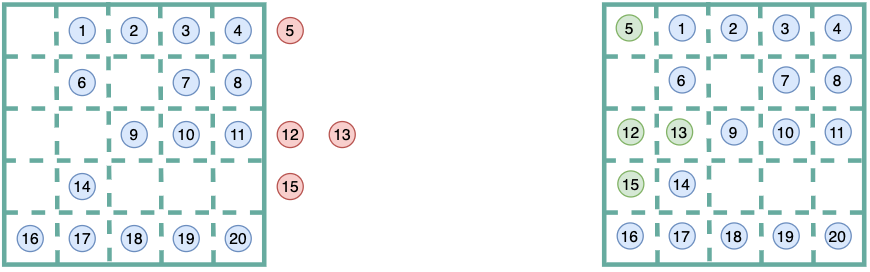
Then, when calculating membrane curvature in mda.Universes that comprises
membrane only (case 1.1), or a membrane with a fixed protein (case 1.2.1),
we should derive surfaces using atoms of reference that fall in the
same unit cell. Here, we manage get those “extra” atoms from out of the
simulation box, back in the same primary unit cell via
MDAnalysis.transformations.wrap.
By wrapping coordinates and keeping all our atoms of reference, we will guarantee two important features in our calculation of membrane curvature:
- More atoms of reference in the primary unit cell. In other words, the
AtomGroupof reference used to derive the surface will have moreatoms, and thefore the surface will be derived from a higher number of points. - As a consequence, our grid will be more populated. Less empty cells in the
grid means we will avoid annoying
np.nansand our gradients (curvature) won’t be driven by undefined values.
Altogether, we will improve our sampling!
These considerations have been brought up during the development of the
MDAnalysis MembraneCurvature tool thanks to the active discussions with my
group of mentors. In the next post, I will discuss in detail the considerations
for the systems with proteins diffusing in the membrane.
Stay tuned for the second part!
References:
- [1] Bloom M, Evans E(1991) Q Rev Biophys 24: 293–397.
- [2] Alberts B. The Cell: A Molecular Approach. 2nd edition.