Membrane Curvature from MD Simulations: Considerations [Part II]
08 August 2021
Considerations to calculate membrane curvature from MD simulations with no position restraints.
In the previous blog post, we summarized the most common Molecular Dynamics setups, and highlighted some points to calculate membrane curvature in two types of simulation systems:
- Membrane only (1.1), and
- Membrane-protein systems, with positions restraints applied on the protein (1.2.1).
In the second part of this series, I will discuss some points to consider to calculate membrane curvature from MD simulations in setups where position restraints are not imposed on proteins.
Position restraints (hereinafter referred as posres) is an algorithm intended to keep particles at a fixed reference position in the simulation box. Although the use of posres in MD is system-dependent and it responds to particular research interests, application of posres is commonly used avoid significant rearrangement of particles. Some of the most common uses are:
- applied to solvent particles to equilibrate around the protein without affecting the protein structure.
- applied to the proteins to keep them fixed in a given position in the membrane.
- applied to lipid headgrops to restrain the oscillation of bilayers to avoid membrane fluctuations.
Given that experimental evidence supports a link between protein diffusion and membrane curvature, and applying posres on the protein prevents protein diffusion, in some cases, it may be a good idea to set MD simulations up with no posres applied on the protein. And, of course, no posres on lipid head groups either :-)
Considerations (Part II)
In MD simulations with no posres, we allow both oscillation of the lipid headgroups as well as protein difussion. Under these settings, the protein is allowed to freely diffuse in the plane of the membrane, and it may subjected to translations and rotations. An example of rotational and translational changes that a protein undergoes while it diffuses is shown in the sequence below:
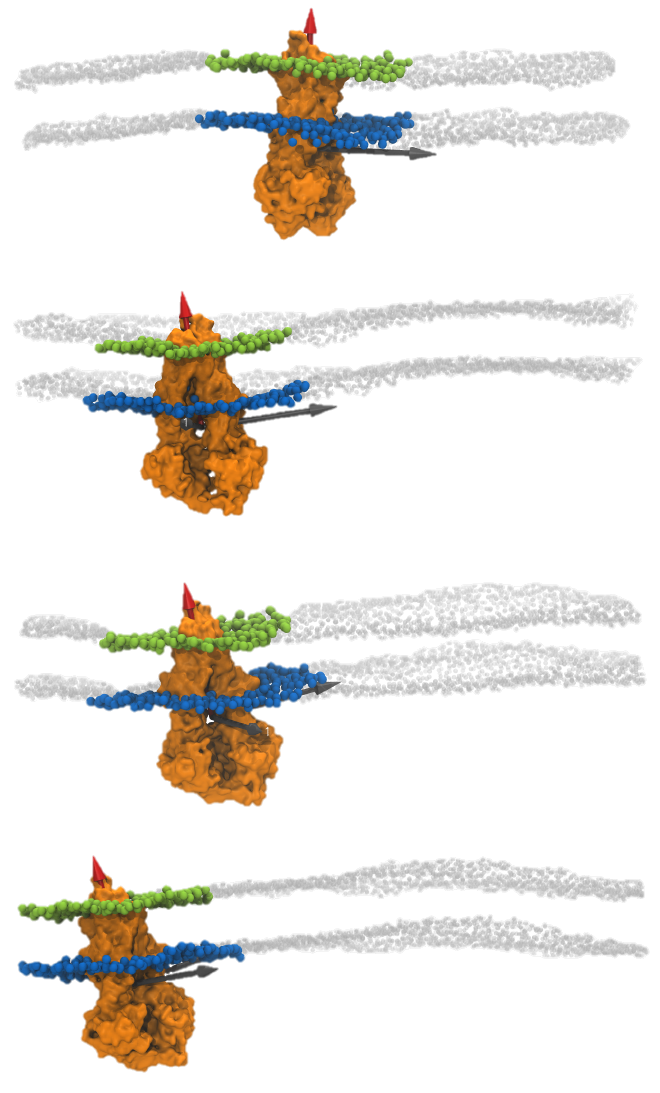
Since the scientific question behind calculating membrane curvature in
membrane-protein systems is what is the membrane curvature induced by the protein, the
trajectory obtained as a result of our MD simulations requires some additional
processing. In this way we guarantee that the calculated curvature is
relative to the protein, and therefore, can be associated to the curvature
induced by the protein.
In the sections below, I am going to dicuss two possible solutions to calculate membrane curvature in systems with membrane-protein systems with no posres applied.
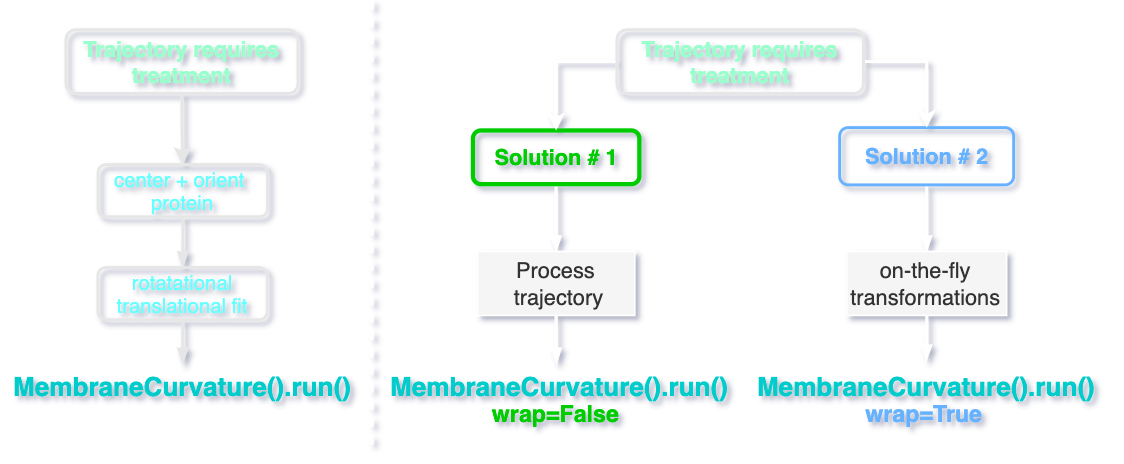
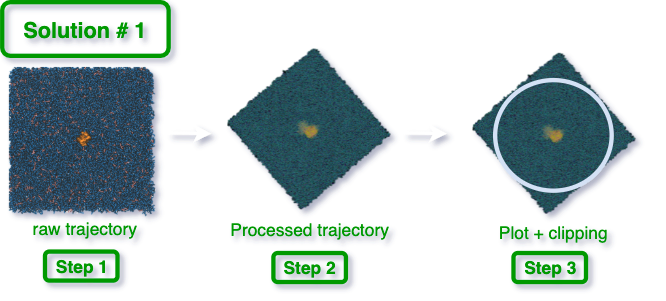
Option 1: Additional trajectory processing
Since the trajectories obtained from MD simulations where proteins dynamics display translational and rotational diffusion need a special treatment, we fundamentally need to perform two main operations. First, we need to center and orient the protein in the center of the simulation box. Then, we need to rotate and translate the lipids, with the protein in the center of the simulation box as a reference.
We can process the trajectory using Gromacs with the commands:
gmx trjconv -pbc whole -ur compact -c
gmx trjconv -fit rot+transxy
With this 2-step trajectory processing, we rotate and translate the lipids of the membrane, while keeping the protein centered in the simulation box.
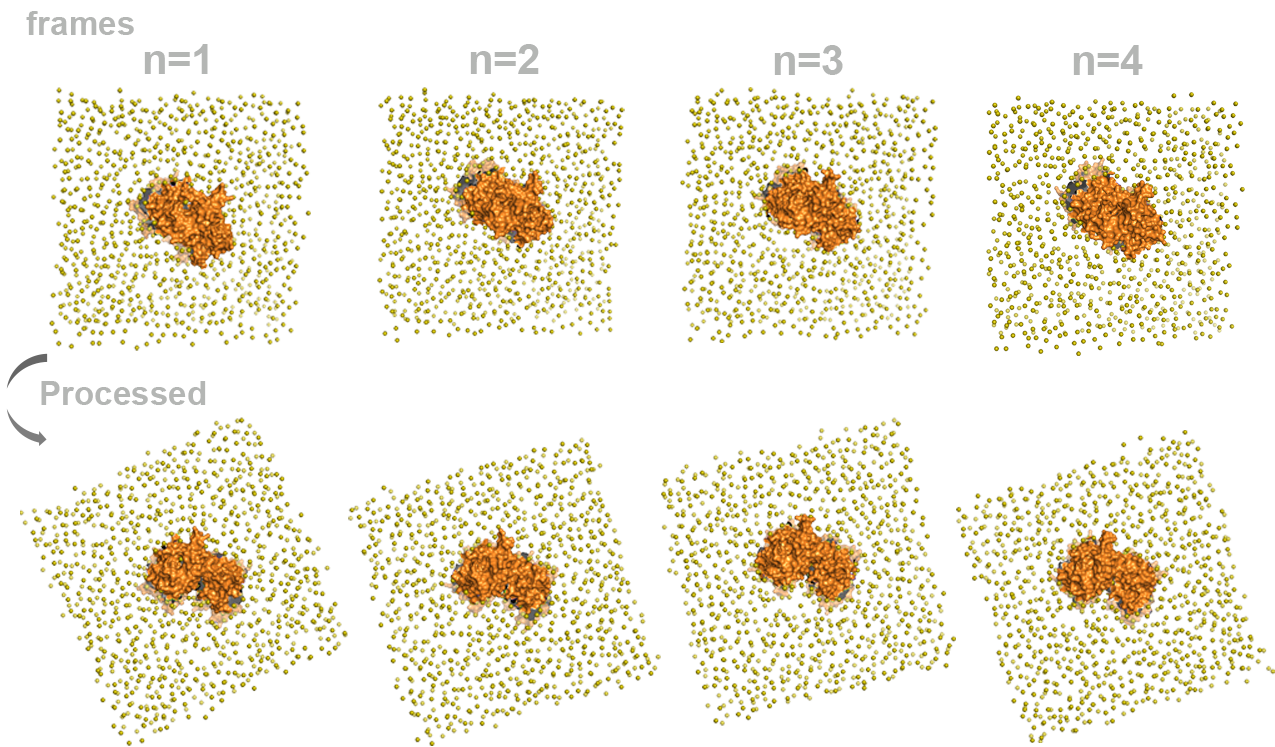
For illustration purposes, I made this short video that shows the difference between the raw trajectory and the processed fit trajectory from the top view of the simulation box. The protein is show in surface representation, and the lipid head groups are shown in spheres.
The snapshots from the video allow us to identify that in the processed trajectory, the structure of the protein remains centered and fixed.
It is worth mentioning that when the trajectory is processed in Gromacs, the
-pbc whole option applies coordinate wrapping, and therefore,
MembraneCurvature should be run with wrap=False when passing a processed
trajectory.
Under these consideration, the processed trajectory can be used to calculate membrane curvature in our MembraneCurvature MDAnalysis tool. To visualize the results, a final clipping would enhance our curvature plot.
Option 2: On-the-fly transformations
A potential alternative to calculate membrane curvature without relying on third parties to process trajectories is possible by applying MDAnalysis on-the-fly transformations.
With this approach we rotate and translate the lipids in every frame of the trajectory, avoiding further trajectory processing by the user and the calculation of curvature would fully rely on MDAnalysis.
So far, MDAnalysis offers two out of the three functionalities to perform
all on-the-fly-transformations: center_in_box and fit_rot_trans.
The center_in_box transformation reaccommodates all the atoms in the simulation
box, allowing a given AtomGroup to be centered in the unit cell. On the other
hand, the fit_rot_trans transformation enables us to fit the trajectory
using an AtomGroup as reference. The functionality of fit_rot_trans is wide
enough that we can perfom fit on the plane of the membrane if needed. For
example,if the fitting is performed on the plane=xy then the transformation
will behave as -fit rotxy+transxy from Gromacs.
A snippet that includes these two transformations looks like:
import MDAnalysis as mda
from MDAnalysis import transformations
from membrane_curvature.base import MembraneCurvature
from membrane_curvature.tests.datafiles import XTC_MEMBPROT_FIT, GRO_MEMBPROT_FIT
universe = mda.Universe(GRO_MEMBPROT_FIT, XTC_MEMBPROT_FIT)
protein = universe.select_atoms("resid 1-1800")
reference_fit = universe.select_atoms("resid 1-1800 and name BB")
workflow_fit = (transformations.center_in_box(protein, center='mass'),
transformations.fit_rot_trans(protein, reference_fit, plane=''xy))
u.trajectory.add_transformations(*workflow_fit)This blog post has more on-the-fly transformations examples to check out!
Currently, the compact option that puts all atoms at the closest distance from
the center of the box is not available in MDAnalysis, so we need extra steps
which may include calculation of rotational and translational matrices applied
to the surface. We are exploring this and other possible approaches to make of
MembraneCurvature a fully-functional MDAnalysis tool!